
Clinical Research Implementation
Industry-sponsored trials
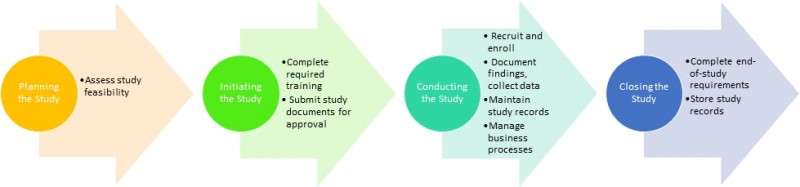
When planning and initiating a study follow these steps: Complete required training, assess study feasibility, submit required documents to the Institutional Review Board and other TU administrative offices (EHS, Biosafety, CoI, etc.), contact the Clinical Research Administration Group to discuss the budget and contractual obligations, fill out the forms (1572, financial disclosure) and other documents, as requested by the sponsor.
When conducting a study follow these steps: Collaborate with site visits, manage your study and maintain the regulatory binder, which consists of the following parts: Clinical Research Protocol, resumes and licensures of key research personnel, logs and documentation, adverse event log, device or drug (also called Investigational Product) log. Be accountable for drug/device use and storage (traceability). Maintain monitoring log, pre-screening log, protocol amendment log/version tracking log , delegation of authority/responsibility log, staff signature log (can be merged with the delegation of authority log), study staff training log, subject contact log, temperature log (if IP is stored in an area other than the investigational pharmacy), tissue log (if applicable). Capture required clinical data in the CRF and start protocol deviation log. Submit invoice to the sponsor, reimburse study participants.
When closing the study follow these steps: Conduct close out visit, notify IRB of study closure, notify other departments of study closure. Consult with legal/public relations department regarding dissemination of results in non-scientific forums and corporate policies. Store records as mandated by the sponsor and obtain final payment.
Investigator-initiated trials (PI)
When planning as study consider the following: Develop a clinical trial protocol and seek design assistance. Consider different experimental designs and control groups. Discuss minimal clinical difference and required samples size. Establish patient recruitment strategy? Assess the time and effort that will be required and estimate the anticipated costs. Identify team members and collaborators. Learn about the regulatory requirements. Set up budget and resource plan. Contact Clinical Research Administration for assistance with budget.
When initiating the study: Complete training required training. Submit required documents to the Institutional Review Board and other TU administrative offices (EHS, Biosafety, CoI, etc.). Register the trial in clinicaltrials.gov.
When conducting a study consider the following: Notify key participants of study start, conduct training (clinical staff, departmental staff) and keep them informed throughout the study. Create a regulatory binder and maintain study documents (see industry-sponsored trials) and set up your data acquisition process (using OnCore or RedCap). Documents study visits in EPIC or in the medical record (inpatients) and reimburse study participants.
When closing the study consider the following: Notify IRB of study closure, notify other departments of study closure, publish clinical research results (is mandatory), close out clinicaltrials.gov info. Consult with media and public relations department regarding dissemination of results in non-scientific forums. Store records for 5 years post study.